Submitting a Monograph
Guidelines for Clinical Verification
Approved July 2020
6. Safety Assurance
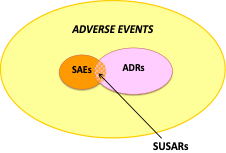
The safety and well-being of human subjects must at all times be held in the highest regard. The FDA and ICH-GCP have established definitions for the various types of adverse experiences or events that may occur in the context of clinical cases or case series, prospective observational studies, or experimental trials. These include Adverse Events (AE), Serious Adverse Events (SAE), Adverse Drug Reactions (ADR), and Suspected Unexpected Serious Adverse Reactions (SUSAR).
Investigators are expected to comply with FDAi and ICH-GCPii guidance on clinical investigations. In addition, investigators shall comply with all requirements of the Ethics Committee or IRB for the study.
- General Requirements
These general requirements and recommendations apply to all permitted research designs and methods (except as noted). Requirements that are applicable to specific research methods are given in separate subsections.
6.1.1. Safety and Reporting during Acquisition of Data
All monograph sponsors must demonstrate an adequate safety and reporting process while acquiring clinical data. The process may vary according to the type of clinical study being conducted.
The safety and reporting process must be evaluated and approved by an Ethics Committee or IRB.
6.1.2. Adverse Events (AE)
An AE is defined as any untoward medical occurrence associated with the use of a drug in humans, whether or not considered drug related.iii
Expected events specific to homeopathy are described in Section 6.1.3. of these guidelines and shall be managed as described in that section.
All shall be reported and managed according to the requirements of the AEs applicable regulations, and Ethics Committee or IRB requirements.
Use of the ‘Adverse Event Determination While Acquiring Clinical Date’ flow chart, located in Section 6.5, is recommended.
6.1.3. Events Specific to Homeopathy
In addition to the FDA/ICH-GCP defined events, there are two expected events specific to homeopathy that can occur during the treatment with an IHMP.
a) Proving Symptoms
Proving Symptoms are expected, mild, transient signs or symptoms occurring within an expected time period after IHMP administration. Such signs or symptoms must be consistent with the current understanding of the homeopathic clinical picture of the IHMP as derived from and supported by prior proving, toxicological, and/or clinical data
b) Homeopathic Aggravations
Homeopathic Aggravations are expected, mild, transient increases of pre-existing signs or symptoms that occur shortly after IHMP administration, quickly resolve, and are associated with an improvement in clinical complaints and / or general health
6.1.4. Recording
The occurrence of either of these types of events given in Section 6.1.3. shall be recorded in the monograph submission as potentially significant to the clinical understanding of the IHMP and form part of the source data of the study.
If, however, either a homeopathic aggravation or proving symptom occurs with an unexpected degree of severity, duration, or frequency, this would also be assessed and managed as an AE (see Flowchart 1 Adverse Event Determination While Acquiring Clinical Data located in Section 6.5).
6.1.5. Causality Assessment
A process for determination of causation likelihood shall be established prior to IHMP administration (for prospective studies) or data analysis (for all studies).
a) Report of causation likelihood shall be included for all AEs using the following labels derived from the FDAiv and ICH guidancev.
i. Possibly related – there is sufficient evidence to suggest a reasonable possibility of a causal relationship between the drug and the AE
ii. Unrelated – there is no evidence to suggest a causal relationship between the drug and the AE
b) Process for determination of causation likelihood must include consideration of all of the following:
i. Temporal relation to IHMP administration
ii. Strength of association
iii. Consistency over repeated dosing (if applicable)
iv. Coherence with known facts on the biology of disease
c) Use of WHO-UMC Causality Assessment System, Naranjo Criteria, Modified FDA algorithm, or similar widely accepted instrument for determination of causality is recommendedvi vii,viii
6.1.6. Adverse Drug Reaction (ADR)
An ADR is any AE that is deemed to be possibly related to the IHMP (see Section 6.1.5 a)).
Therefore, all AEs deemed to be possibly related to the IHMP shall be managed as ADRs. This process must include:
d) Report of causation likelihood
e) Report of AE timeline and outcome using the following categories:
vii. Death: The drug may have contributed to the death
viii. Not recovered/not resolved: The subject has not yet recovered
ix. Recovered/resolved with sequelae: The subject recovered, but with an after effect possibly due to disease, injury, treatment, or procedure
x. Recovered/resolved: The subject fully recovered
xi. Unknown: The reporter did not know the outcome at the time the report was submitted
f) Compliance with all applicable regulatory requirements
6.1.7. Serious Adverse Events (SAE)
SAEs are defined17,ix, x as any untoward medical occurrence, associated with the use of a drug in humans, whether or not considered drug related, that at any dose:
a) Results in death
b) Is life-threatening
c) Requires inpatient hospitalization or prolongation of existing hospitalization
d) Results in persistent or significant disability/incapacity
e) Is a congenital anomaly/birth defect
f) Or requires medical or surgical intervention to prevent hospitalization, permanent impairment or damage
All SAEs must be recorded and reported to the following bodies:
a) Ethics Committee or IRB within the specified time period
b) The manufacturer of the IHMP (within 24 hours for prospective studies)
c) Government agency(s) in the time and manner according to any applicable legal requirements of the country
A pre-determined written protocol shall be followed for reporting and management of SAE’s (prospective studies only).
6.1.8. Suspected Unexpected Serious Adverse Reaction (SUSAR)
A SUSAR is a SAE (see Section 6.1.7), that is suspected to have a causal relationship with the IHMP.
All SUSARS must be recorded and reported to all of the following:
a) Ethics Committee or IRB within the specified time period
b) The manufacturer of the IHMP (within 24 hours for prospective studies)
c) Government agency(ies) in the time and manner according to any applicable legal requirements of the country
Figure 1 delineates the various categories and relationships of AEs. The scale of the figure does not reflect actual frequencies of the various events.
6.1.9. Monograph Reporting
All ADRs, AEs, proving symptoms, and Homeopathic Aggravations shall be included and delineated in the Monograph Report.
If ADRs, AEs, proving symptoms, or Homeopathic Aggravations do not occur during data acquisition, this must be stated.
- Specific Requirements for Clinical Case Series
A process for evaluation and reporting AEs for monograph submission is recommended.
6.2.1. Retrospectively Collected Cases
A specific AE handling process is not required for retrospectively collected cases.
Monograph sponsor shall report what reasonable efforts were made to collect and assess safety information on the IHMP (including AEs, homeopathic specific reactions, and ADRs), despite the inherent challenges with collecting this type of data retrospectively.
6.2.2. Prospectively Collected Cases
Sponsors collecting data from prospective cases shall include a process for AE handling by the healthcare providers involved in the investigational use of the IHMP. This process must follow the requirements listed in Section 6.1.
- Specific Requirements for Prospective Observational Studies
Monograph sponsors shall ensure adherence to all general requirements and specific requirements for prospective observational studies detailed in this section.
6.3.1. Safety Management Plan
Because subjects receive therapy with unknown or unconfirmed therapeutic action, this form of study requires a detailed Safety Management plan with specific personnel designated to manage the plan.
A safety management plan shall contain standard operating procedures (SOPs) for AE handling, treatment if necessary, and reporting.
The plan shall define the process of handling and managing of AEs, including SUSARs and SAEs – taking into account any applicable regulatory requirements with regards to reporting timelines.
For all studies except retrospective case studies, a clear plan for the recording, assessment and reporting, of AEs, approved by the Ethics Committee or IRB, shall form part of the study protocol.
Monograph sponsors are recommended to employ an independent monitor for the study to ensure that the rights and well-being of subjects are protected, and the study data is accurately reported and verifiable.
- Specific Requirements for Experimental Trials
Monograph sponsors must ensure adherence to all general requirements (Section 6.1), specific requirements for prospective observational studies (Section 6.3), and specific requirements for experimental trials detailed below.
6.4.1. Trial Monitoring
Trial monitoring is required to conform to the ICH-GCP guidelines.
Monograph sponsors shall submit a detailed report of the monitoring process with any controlled trial data.
6.4.2. Procedure for Un-blinding in Relation to Adverse Events
For experimental trials with blinded allocation, a system for un-blinding of a single subject status in relation to an AE shall be established prior to trial initiation.
Requirements for procedures un-blinding are given in Section 5.4.3.
6.4.3. Individual Subject Discontinuation from a Study
All subjects with an AE, where the therapeutic intervention related to the IHMP that has been determined by the PI to potentially affect the clinical outcome, safety, or well-being of the subject, shall be discontinued from the experimental trial.
Requirements for procedures subject discontinuation are given in Section 5.4.5.
6.4.4. Premature Termination of a Trial
If a trial is prematurely discontinued, the Ethics Committee or IRB, monograph sponsor, and any relevant regulatory authorities must be notified promptly including the reason for termination or suspension of the trial.
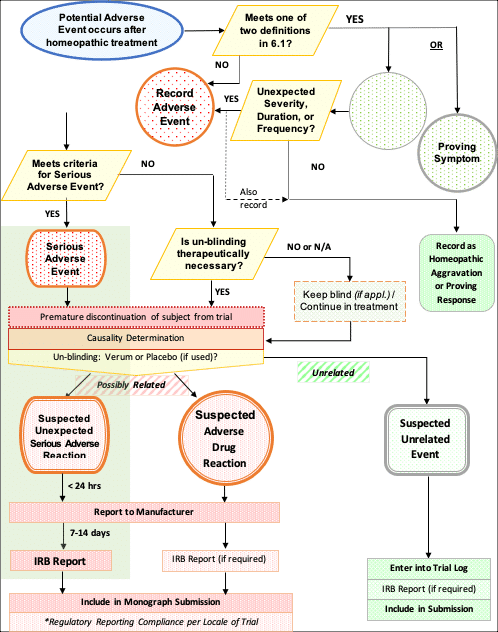
- Adverse Event Determination While Acquiring Clinical Data
Flowchart 1: Adverse Event Determination While Acquiring Clinical Data
i) U.S. Food and Drug Administration (FDA). Guidance Documents (Including Information Sheets) and Notices. FDA website. https://www.fda.gov/science-research/clinical-trials-and-human-subject-protection/guidance-documents-including-information-sheets-and-notices. Content current as of 03/30/2018. Accessed September 10, 2020.
ii) International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). E2B(R3) Individual Case Safety Report (ICSR) Specification and Related Files. http://www.ich.org/fileadmin/Public_https://ich.org/page/e2br3-individual-case-safety-report-icsr-specification-and-related-files. Last updated: August, 2020. Accessed September 10, 2020.
iii) U.S. Code of Federal Regulations: Title 21—Food and Drugs: Chapter I—Food and Drug Administration, Department of Health and Human Services: Subchapter D—Drugs for Human Use: Part 312—Investigational New Drug Application: Subpart B—Investigational New Drug Application (IND): §312.32 IND safety reporting: (a) Definitions. Electronic Code of Federal Regulations (e-CFR) website. https://www.ecfr.gov/cgi-bin/text-idx?SID=d1517142cff82c2bad491c77a1d7e8fa&mc=true&node=pt21.5.312&rgn=div5#se21.5.312_132. e-CFR data is current as of September 8, 2020. Accessed September 10, 2020.
iv) U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Guidance for Industry and Investigators. Safety Reporting Requirements for INDs and BA/BE. https://www.fda.gov/media/79394/download. Issued December 2012. Accessed September 10, 2020.
v) International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). ICH Harmonised Tripartite Guideline: Clinical Safety Data Management: Definitions and Standards for Expedited Reporting: E2A. Current Step 4 version dated 27 October 1994. https://database.ich.org/sites/default/files/E2A_Guideline.pdf. Dated 27 October 1994. Accessed September 10, 2020.
vi) World Health Organization (WHO). WHO Guidelines on Safety Monitoring of Herbal Medicines in Pharmacovigilance Systems. Geneva, Switzerland: World Health Organization, 2004. https://apps.who.int/iris/handle/10665/43034. Accessed September 10, 2020.
vii) Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981,30(2):239-245. Doi: 10.1038/clpt.1981.154
viii) Ide K, Yamada H, Kitagawa M, et al. Methods for estimating causal relationships of adverse events with dietary supplements. BMJ Open 2015, 5(11):e009038:1–6. Doi: 10.1136/bmjopen-2015-009038. https://bmjopen.bmj.com/content/bmjopen/5/11/e009038.full.pdf. Accessed September 10, 2020.
ix) U.S. Food and Drug Administration (FDA). MedWatch: The FDA Safety Information and Adverse Event Reporting Program. Reporting Serious Problem to FDA: What is a serious adverse event? FDA website. https://www.fda.gov/safety/reporting-serious-problems-fda/what-serious-adverse-event. Content current as of: 02/01/2016. Accessed September 10, 2020.
x) U.S. Department of Health and Human Services, U.S. Food and Drug Administration. 21 CFR Parts 312 and 320 [Docket No. FDA–2000–N–0108] (formerly Docket No. 00N–1484): RIN 0910–AG13: Investigational New Drug Safety Reporting Requirements for Human Drug and Biological Products and Safety Reporting Requirements for Bioavailability and Bioequivalence Studies in Humans. Fed. Regist. 2010;75(188):59935-59963. http://frwebgate.access.gpo.gov/cgi-bin/getdoc.cgi?dbname=2010_register&docid=fr29se10-3.pdf. Accessed September 10, 2020.