Submitting a Monograph
Guidelines for Homeopathic Drug Provings
Approved April 2015
6. Safety Assurance
FDA and ICH-GCP have established guidelines for adverse event definitions for clinical trials in humans. Adverse Events (AEs) are any untoward medical occurrence in subjects who are administered a pharmaceutical product during a clinical trial, irrespective of whether there is a causal relationship with the product.
Subjects in Provings are expected to develop a range of transient symptoms when taking the Investigational Proving Substance (IPS). Such symptoms are essential to the evaluation and description of clinical usefulness of the IPS. Because of the investigatory nature of Provings with respect to new medicinal substances being tested, the exact range of symptom type and severity may not be predicted prior to Proving initiation.
Within the context of Provings, any symptom or condition that occurs in a test subject during the Proving and is clinically unexpected is considered an AE. If an AE fulfills at least one of the criteria listed in Section 6.3 below, it is defined as a Serious Adverse Event (SAE). The establishment of appropriate protocol definitions and procedures will ensure the appropriate identification and handling of AEs, while avoiding inappropriate reporting of Proving symptoms as AEs. For a visual representation, see the Figure 1 below:
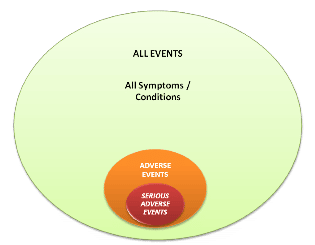
A further aspect of AE reporting is the assessment of the possible categorization as an Adverse Drug Reaction. If a causal relationship between the IPS and the AE is at least a reasonable possibility, the AE becomes an Adverse Drug Reaction. And if the causal relationship between the IPS and the SAE is at least a reasonable possibility, then the SAE becomes a Suspected Unexpected Serious Adverse Reaction (SUSAR).
- Adverse Events
Any noxious, unintended, or untoward medical occurrence that may appear or worsen in a subject during the course of a proving and which is unexpected and clinically significant should be considered an Adverse Event.
Unexpected symptoms and signs have at least one of the following characteristics:
a) have duration longer than the proving period
b) have clinical severity greater than described in the Informed Consent
c) have clinical severity that falls within the definition of Serious Adverse Event
d) require medically necessary therapeutic intervention,
e) results in removal from the Proving
All AEs should be reported and managed according to the requirements of all applicable regulations and Ethics and Institutional Board Requirements.
A process for determination of likelihood of causation should be established prior to IPS administration.
It is recommended that the possibility of causation be determined prior to un-blinding and based upon the following factors(6):
a) Temporal relation to IPS administration
b) Strength of association
c) Consistency over repeated dosing within one subject or across the study population
d) Observed increase of symptoms with repetition of dose or increase in attenuation (if any in the design)
e) Coherence with known facts on the biology of disease
f) Once identity of IPS is known, causality may be reassessed based upon known biological properties of the IPS (if feasible, should be done before un-blinding as to verum or placebo allocation)
Report of causation likelihood should be included for all AEs using the following labels derived from the FDA and ICH.(1)
a) ‘Possibly related’ meaning there is evidence to suggest a ‘Reasonable possibility’ of a causal relationship between the drug and the adverse event.
b) ‘Unrelated’ meaning there is no evidence to suggest a causal relationship between the drug and the adverse event.
All AEs with a possible causal relationship to the IPS should be reported and managed as possible Adverse Drug Reactions, including:
a) Report to Pharmacopœia Revision Committee (PRC) the following information using standard reporting categorization provided in the International Conference on Harmonization tripartite guidance on Clinical Safety Data Management E2Br (R2)(2)
1. Report of causation likelihood
2. Report of adverse event timeline and outcome
i. Death: The drug may have contributed to the death.
ii. Not recovered/not resolved: The subject has not yet recovered.
iii. Recovered/resolved with sequelae: The subject recovered, but with an after effect possibly due to disease, injury, treatment, or procedure.
iv. Recovered/resolved: The subject fully recovered.
v. Unknown: The reporter did not know the outcome at the time the report was submitted.
b) Adverse Drug Reactions that potentially change the risk profile need to be reported to the relevant Competent Authority (e.g. FDA) and to the IRB in accordance with applicable regulations. Unanticipated adverse events which create a problem for the conduct of the trial should be reported to the IRB(1)
c) Compliance with any applicable regulations
d) Reporting to the Manufacturer.
- Serious Adverse Events
Serious adverse events are defined by the FDA (3), (4), (5) as any untoward medical occurrence that at any dose:
a) Results in death
b) Is life-threatening
c) Requires inpatient hospitalization or prolongation of existing hospitalization
d) Results in persistent or significant disability/incapacity
e) Is a congenital anomaly/birth defect
f) Or, requires medical or surgical intervention to prevent hospitalization, permanent impairment or damage
All serious adverse events must be recorded and reported:
a) To Ethics or Institutional Review board within the specified time period
b) To the Manufacturer of the IPS within 24 hours
c) And to government agency(s) in the time and manner according to any applicable legal requirements of the country(s) where the Proving is being conducted
A pre-determined written protocol should be followed for reporting and management of serious adverse events during the Proving.
- Single Subject Un-blinding in Connection with Adverse Events
A system for un-blinding of a single subject status in relation to an adverse event should be established prior to Proving initiation.
Decision to un-blind an individual Proving subject in the case of an AE should be based upon therapeutic necessity.
SAEs may require un-blinding at the time of the event.
Un-blinding protocol should comply with the following requirements:
a) Maintains blinding during the Proving unless un-blinding is required;
b) Limits the number of personnel within Proving team who will have access to un-blinding information to those absolutely necessary;
c) Controls which personnel have authority to access data;
d) If individual blind is broken, records any data access including specific personnel who obtained or viewed this information, information that was obtained, date in which it was obtained, and reason for un-blinding;
e) Includes protocol for when un-blinding should occur;
f) And contains adequate “fail-safe” procedures to assure immediate access to subject data during the Proving.
- Individual Subject Discontinuation from Proving
All subjects with an AE, where the therapeutic intervention has been determined by the Investigator to potentially affect the Proving, should be discontinued from the Proving. A complete record of the discontinuance and reasons should be part of the study record.
If subject is withdrawn/ discontinued due to symptom reliability concerns, no further data from that subject should be included in the Proving analysis.
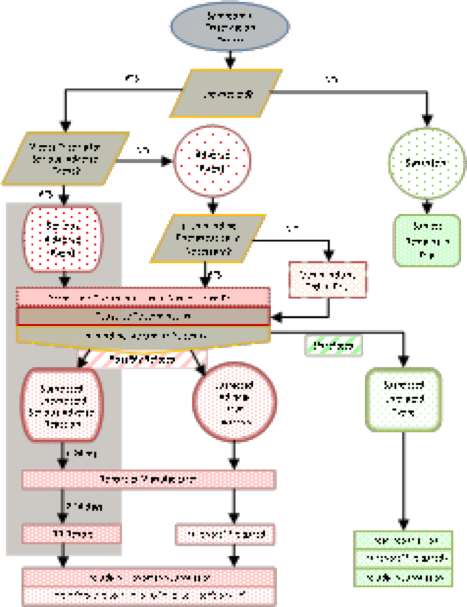
- Event Handling Flow Chart
A flow chart for new symptom assessment to guide Study Personnel in the recognition and management of suspected adverse drug reactions (including SUSARs) should be developed. Chart 1 (below) may be used as a template.
The proposed timeframes associated with SAE reporting are recommended; variations based on local requirements may apply.
Flow chart for adverse event determination in a proving
(1) United States Food and Drug Administration. Guidance for Industry and Investigators. Safety Reporting Requirements for INDs and BA/BE. 21CFR312.32. Revised April 01, 2011. Last downloaded April 01, 2012 from online at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM227351.pdf
(2) ICH Harmonized Tripartite Guideline Maintenance of the ICH Guideline on Clinical Safety Data Management: Data Elements For Transmission Of Individual Case Safety Reports E2B(R2) Current Step 4 version dated 5 February 2001. Last downloaded April 01, 2012 from online at
http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E2B/Step4/E2B_R2__Guideline.pdf
(3) United States Food and Drug Administration MedWatch: The FDA Safety Information and Adverse Event Reporting Program. Last downloaded April 01, 2012 from online at
http://www.fda.gov/safety/medwatch/howtoreport/ucm053087.htm
(4) Department Of Health And Human Services Food and Drug Administration 21 CFR Parts 312 and 320 [Doc